Ежегодно у небольшой части пациентов с ХЛЛ/ЛМЛ развивается диффузная В-крупноклеточная лимфома или лимфома Ходжкина. Появление крупноклеточной лимфомы на фоне ХЛЛ называется синдромом Рихтера. Появление лимфомы Ходжкина на фоне ХЛЛ называется ходжкинской трансформацией. Отличительной особенностью клеток при таких трансформациях является сохранение исходного фенотипа ХЛЛ.
Синдром Рихтера встречается примерно у 3–5 % пациентов с ХЛЛ в течение заболевания, со скоростью трансформации от 0.5% до 1% в год.
Общие сведения о синдроме Рихтера
Первое описание трансформации было предложено Maurice Richter в его статье 1928 года, описывающей патологический вид «генерализованной ретикулярно-клеточной саркомы» у пациента с ХЛЛ. Сорок лет спустя Lortholary и др. ввели термин «синдром Рихтера» для описания диффузной В-круноклеточной лимфомы (ДВКЛ), возникающей у пациентов с ХЛЛ. Международная рабочая группа по изучению ХЛЛ (IWCLL) определяет синдром Рихтера как трансформацию ХЛЛ в более агрессивную лимфому.
Подавляющее большинство таких преобразований представляют собой ДВКЛ. Гораздо реже может возникать трансформация ХЛЛ в лимфому Ходжкина (ходжкинская трансформация). Согласно алгоритму Hans-Choi (основанному на иммуногистохимии), от 90% до 95% трансформаций ДВКЛ у пациентов с ХЛЛ относятся к более агрессивному активированному подтипу B-клеток.
Проведенные исследования разделили ДВКЛ на фоне ХЛЛ на 2 типа:
– ДВКЛ «клонально связанную» с ХЛЛ (~80% случаев)
– ДВКЛ «клонально не связанную» с ХЛЛ (~20% случаев).
Различие между этими типами ДВКЛ имеет решающее значение как для прогноза, так и для лечения. Медиана выживаемости пациентов при «клонально связанной» ДВКЛ составляет примерно 1 год. Прогноз у пациентов с «клонально не связанной» ДВКЛ аналогичен прогнозу для самостоятельной, вновь выявленной ДВКЛ .
Сочетание герминальных генетических характеристик, клинических особенностей (поздняя стадия лейкоза), биологических (немутированный статус IGHV, стереотипные антигенные рецепторы, ZAP-70 + , CD38 + , CD49d +) и цитогенетических (del17p13.1 или del11q23.1) характеристик В-клеток ХЛЛ, а также некоторых методов лечения ХЛЛ, связаны с более высоким риском синдрома Рихтера. Недавние исследования также выявили решающую роль потери CDKN2A, мутаций TP53, активации C-MYC и мутаций NOTCH1 в возникновении синдрома Рихтера.
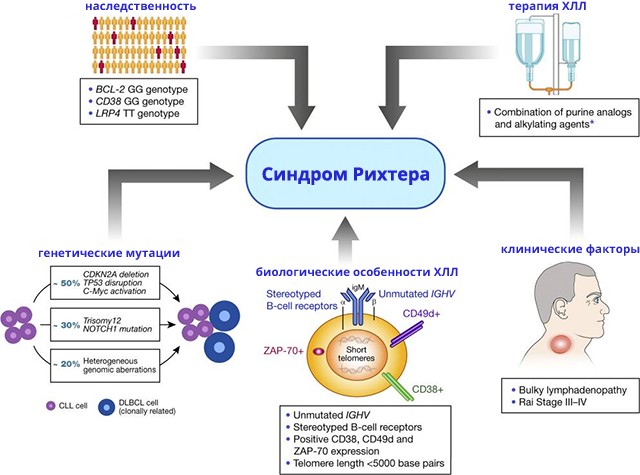
Наследственная предрасположенность
Наследственные полиморфизмы герминальной линии могут предрасполагать некоторых людей, у которых развился ХЛЛ, к последующей трансформации. В частности, сообщалось, что полиморфизмы герминальной линии в генах В-клеточной лимфомы 2 (BCL-2), CD38 и белка LRP4, связанного с рецептором липопротеинов низкой плотности, повышают риск развития СР.
Клинико-лабораторные особенности при диагностике ХЛЛ
Расширенная стадия Rai (III-IV) при диагностике ХЛЛ и лимфатические узлы >3 см при физикальном осмотре являются единственными клиническими признаками, связанными с более высоким риском развития синдрома Рихтера в будущем.
Биологические характеристики В-клетки ХЛЛ
Некоторые характеристики рецептора B-клеток тесно связаны с СР. Пациенты с ХЛЛ с лейкемическими В-клетками, не мутировавшими по IGHV, имеют примерно 4-кратный риск СР по сравнению с пациентами с мутацией IGHV.
Примерно 30% всех пациентов с ХЛЛ имеют высокую степень сходства в структуре B-клеточных рецепторов из-за гомологии CDR3, определяющей комплементарность тяжелой цепи. Эти пациенты с стереотипными B-клеточными рецепторами имеют более высокий риск развития синдрома Рихтера, независимо от статуса мутации IGHV. В некоторых исследованиях сообщалось, что использование семейства IGHV4-39 связано с более высоким риском СР.
Соматические генетические характеристики
Ранние исследования показали, что генетические аномалии del(11q23.1) и del(17p13.1), выявляемые с помощью FISH, связаны с возникновением СР. Более поздние исследования показывают, что пациенты с ХЛЛ с мутацией NOTCH1, имеют значительно более высокую вероятность трансформации (45%) по сравнению с пациентами без этого дефекта (4%), что указывает на важную роль этого лиганд-зависимого фактора транскрипции в патогенезе СР. Также отмечено, что более короткая длина теломер, которая является маркером генетической нестабильности, связана с повышенным риском возникновения синдрома Рихтера.
Терапия ХЛЛ и риск трансформации
В когортном исследовании 1641 пациента с впервые диагностированным ХЛЛ, наблюдавшегося в клинике Mayo и проспективного наблюдения, скорость трансформации увеличилась с 0,5% в год до терапии до 1% в год после лечения ХЛЛ. В частности, риск возникновения синдрома Рихтера увеличился в 3 раза среди пациентов, которые получали комбинацию алкилирующих агентов (которые вызывают повреждение ДНК) и аналогов пуринов (которые нарушают репарацию ДНК), но не увеличивается для тех, кто получал только один из этих классов препаратов.
Хотя таргетные препараты, такие как Ибрутиниб и Иделалисиб, продемонстрировали значительную эффективность при ХЛЛ, мало что известно об их связи с возникновением СР. В отчете о 85 пациентах с рецидивирующим/рефрактерным ХЛЛ, получавших ибрутиниб и получавших ранее тяжелое лечение, у 7 (8%) пациентов развился СР.
Иммуносупрессия и РС
Инфицирование вирусом Эпштейна-Барр (ВЭБ) было зарегистрировано в 0–15% образцов тканей у пациентов с СР. Является ли ВЭБ причиной СР у этих пациентов или он является просто проявлением основного иммуносупрессивного состояния, связанного с ХЛЛ, до конца не понятно.
Диагностика синдрома Рихтера
Некоторые признаки и симптомы, хотя и неспецифичны, должны предупредить врача о возможной трансформации. К ним относятся высокая температура, быстрое увеличение лимфатических узлов, потеря веса, гиперкальциемия и заметно повышенный уровень ЛДГ. Иногда экстранодальное поражение центральной нервной системы, глаз, яичек и легких также может быть проявлением СР. Хотя они вызывают подозрение на СР, все эти признаки и симптомы также могут быть следствием прогрессирующего ХЛЛ, особенно после возникновения делеции 17p13.1 или мутаций в гене TP53.
Пациентам с подозрением на ХЛЛ/ЛМЛ либо с установленным диагнозом ХЛЛ/ЛМЛ, у которых имеются клинические признаки синдрома Рихтера или ходжкинской трансформации (прогрессирующий рост лимфатических узлов, потеря веса, персистирующая лихорадка без признаков инфекции, высокий уровень лактатдегидрогеназы, гиперкальциемия), рекомендуется выполнить позитронную эмиссионную томографию (ПЭТ) с применением 18F-фтордезоксиглюкозы для выявления зон с вероятной трансформацией и выбора оптимального очага для проведения биопсии.
При значениях SUV <5 вероятность синдрома Рихтера составляет около 3 %. Для синдрома Рихтера типичны значения SUV >15. Если интенсивность накопления препарата отчетливо различается в разных зонах, необходимо выполнение биопсии наиболее активного узла или очага. Синдром Рихтера констатируется только на основании гистологического исследования.
Определение клонального родства относительно просто в подгруппе пациентов, у которых ткань ХЛЛ и ДВКЛ имеет различную рестрикцию легких цепей иммуноглобулина (только Kappa или только Lambda). Для большинства пациентов, у которых рестрикция легких цепей одинакова (есть и Kappa и Lambda), для определения клонального родства необходимо секвенирование гена IGHV как исходного ХЛЛ, так и ткани ДВКЛ. В клонально родственных случаях СР последовательность IGHV VDJ будет идентична (или почти идентична) между ДВКЛ и исходной тканью ХЛЛ.
Также, экспрессия белка LEF1 (ядерная экспрессия) характерна для лимфоцитарной лимфомы с трансформацией в диффузную В-клеточную крупноклеточную лимфому и позволяет провести дифференциальную диагностику с CD5+ диффузной В-клеточной крупноклеточной лимфомой.
Прогноз при синдроме Рихтера
Наиболее важным фактором, определяющим клинический исход у пациентов с СР, является клональная связь ДВКЛ с основным ХЛЛ. Пациенты с клонально несвязанным ДВКЛ имеют медианную выживаемость примерно 62 месяца, что сопоставимо с пациентами с ДВКЛ de novo. С другой стороны, медиана выживаемости пациентов с клонально связанным СР значительно короче и составляет от 8 до 14 месяцев.
Варианты терапии при синдроме Рихтера
Для лечения пациентов с синдромом Рихтера на фоне ХЛЛ традиционно используют схемы на основе антрациклинов (ритуксимаб, циклофосфамид, доксорубицин, винкристин и преднизолон [режим R-CHOP]). Такие режимы являются стандартным лечением ДВКЛ у пациентов с гистологически подтвержденной трансформацией в более агрессивную лимфому, т.е. с клонально связанной ДВКЛ.
Также, согласно недавним исследованиям BRUIN (NCT03740529 ), нековалентный ингибитор BTK Пиртобрутиниб продемонстрировал хорошую эффективность и переносимость у пациентов с рецидивирующими или рефрактерными В-клеточными злокачественными новообразованиями, включая тех, у кого прогрессирование зафиксировано на фоне приема ковалентных ингибиторов BTK, таких как ибрутиниб и акалабрутиниб.
В случаях с клонально несвязанной ДВКЛ терапия производится по стандартам Клинических рекомендаций по лечению “Агрессивных нефолликулярных лимфом ” (Минздрава России, для Российских пациентов).
При подготовке данной статьи использованы:
1. Parikh SA, Kay NE, Shanafelt TD. “How we treat Richter syndrome “. Blood. 2014 Mar 13;123(11):1647-57.
2. Клинические рекомендации по лечению ХЛЛ/ЛМЛ. // «Российское общество онкогематологов», И.В. Поддубная; «Национальное гематологическое общество», Е.Н. Паровичникова; «Ассоциация онкологов России», А.Д. Каприн. РФ, 2022 год.