На момент установления диагноза ХЛЛ частота выявления делеции короткого плеча хромосомы 17 (делеция 17р) у больных варьирует в пределах 3-8%. У пациентов с прогрессирующим и рефрактерным ХЛЛ она может достигать 30%.
Делеция 17p13 приводит к потере локуса гена TP53 в одном аллеле, а в примерно 80% случаев делеция TP53 сочетается с мутацией гена TP53 во втором аллеле. Некоторые мутации гена TP53 могут происходить вообще без делеции 17p, но это встречается довольно редко – в 5% случаев.
По современным нормам, выполнение только одного FISH на делецию 17p13 недостаточно. Для того чтобы идентифицировать наибольшее число пациентов с устойчивостью к ИХТ, необходимо также исследовать мутации TP53. Наличие мутации констатируется, если число клеток с мутацией превышает 10 %.
На что влияют поломки 17р/ТР53?
Ген ТР53 является супрессором опухолевого роста. Важнейшей функцией белка ТР53 является подавление роста генетически поврежденных, соответственно, потенциально опухолевых клеток. На TP53 сходятся многочисленные сигналы, отслеживающие состояние клетки и ее окружения. Это достигается за счет разного рода модификаций белковой р53 молекулы посредством фосфорилирования, ацетилирования, связывания с другими молекулами, регулирующими его активность.
В результате действия механизмов, природа которых в настоящее время не ясна, повреждения ДНК в норме стимулируют синтез белка ТР53. Это в свою очередь “запускает” продукцию белковых молекул: р21, ингибиующих циклин-зависимые киназы, GADD45, тормозящих клеточный рост; ERCC, распознающих и вырезающих поврежденные участки ДНК. Данные молекулы участвуют в задержке роста и деления клеток во время восстановления структуры ДНК. ТР53 элиминирует поврежденные клетки из популяции, таким образом препятствуя репликации ДНК до репарации повреждения. При необратимом повреждении ДНК индуцируется апоптоз.
Больные с делецией 17р13 и мутациями в ТР53 характеризуются низкой ОВ и быстрой прогрессией заболевания. Тем не менее, недавние исследования показали клиническую неоднородность у пациентов с делецией 17p в зависимости от времени возникновения этой аномалии: как раннего события или более частого вторичного изменения. Больные с выявленной делецией 17p в дебюте заболевания имеют более продолжительную ОВ (4-5 лет) в отличие от больных с делецией 17p, приобретенной на этапе прогрессии заболевания (1-1,5 года).
В большинстве случаев мутации в гене ТР5З сочетаются с немутированным IGHV. Однако можно выделить небольшую подгруппу пациентов с ХЛЛ, у которых мутации в ТР5З сочетаются с мутированным IGHV. У таких пациентов течение ХЛЛ может быть индолентным, а необходимость в терапии может вовсе не возникнуть. Таким образом, выявление самой по себе мутации в ТР5З нельзя рассматривать как однозначный фактор неблагоприятного прогноза.
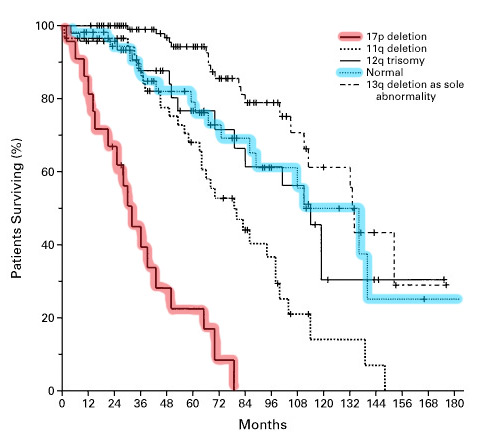
Голубая линия – мутаций нет, красная – делеция 17p.
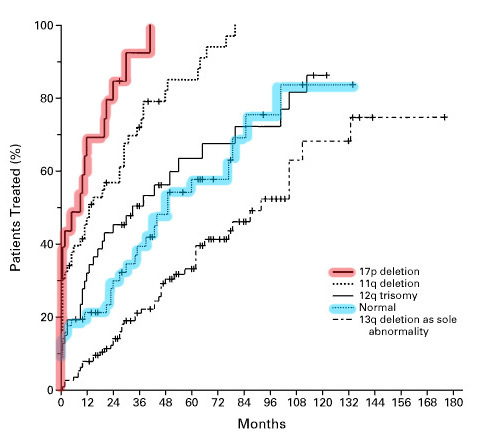
Голубая линия – мутаций нет, красная – делеция 17p.
Как лечат пациентов с поломками 17р/ТР53?
Показано, что делеция 17р13 достоверно чаще встречается у больных с немутированным вариантом генов IGHV. Пациенты с выявленной делецией 17p и/или мутациями TP53 достоверно хуже отвечают на ИХТ стандартными протоколами FCR (флударабин, циклофосфамид, ритуксимаб) и BR (бендамустин, ритуксимаб). У таких больных часто выявляется резистентность к флударабину, полные ремиссии наступают редко и они непродолжительны.
Включение в схему терапии новых таргетных препаратов таких, как ингибиторы тирозинкиназы Брутона (Ибрутиниб, Акалабрутниб) и ингибиторы bcl-2 (Венетоклакc), значительно улучшает показатели ОВ и ВБП. У молодых пациентов, при возможности, терапия Ибрутинибом должна служить подготовкой к трансплантации аллогенных стволовых клеток.
При подготовке этой статьи использованы материалы:
1. Клинические рекомендации “Хронический лимфоцитарный лейкоз / лимфома из малых лимфоцитов”, ID:134. Минздрав РФ, Москва, 2020 год.
2. “Характеристика кариотипа иммуностимулированных В-лимфоцитов больных ХЛЛ”. Кислицына М.А., ФГБУ “НМИЦ Гематологии” Минздрава РФ, Москва, 2021 год.
3. “Genomic aberrations and survival in chronic lymphocytic leukemia”. H. Dohner, S. Stilgenbauer, A. Benner, E. Leupolt, A. Krober, L. Bullinger, K. Dohner, M. Bentz, P. Lichter // N. Engl. J. Med. – 2000. – Т. 343 – №26 – 1910–1916с.